COVID-19感染細胞のトランスクリプトーム解析
概要
ヒトの肺モデル細胞にウィルスを感染させ、それぞれmRNAの発現量の計測した (Blanco-Melo et al. 2020)。公開されているデータを用いて発現差異解析を行う。
| cell line | |
|---|---|
| NHBE | 正常ヒト気管支上皮細胞 |
| A549 | ヒト肺胞基底上皮腺癌細胞 |
| Calu3 | ヒト肺気道がん細胞 |
| A549-ACE2 | A549でACE2を過剰発現させた株 |
| virus | |
|---|---|
| Mock | 感染なし(コントロール) |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| IAV | influenza A virus |
| RSV | human respiratory syncytial virus |
| HPIV3 | Human parainfluenza viruses 3 |
| IAVdNS1 | influenza A virus, NS1欠損株 |
| SARS-CoV-2_Rux | SARS-CoV-2 + Ruxolitinib |
以下では、NHBE(正常ヒト気管支上皮細胞)にSARS-CoV-2を感染させたサンプルについてコントロールとの発現差異を検出する例を示す。 上記の公開データでは他にも様々な条件で計測したデータが提供されているので、各自様々な条件における発現差異を検出し、機能解析を試されたい。
データのダウンロード
- http://usegalaxy.eu/ にログインする。
- 新しいヒストリーを作成し、「COVID-19: transcriptional response」という名前を付ける。
- データをアップロードする。
- 左上のアップロードボタンを押す。
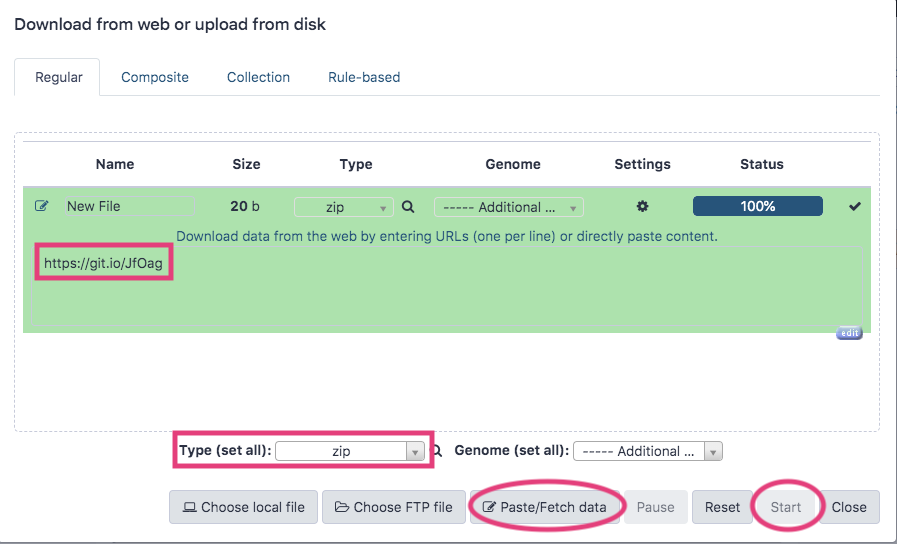
- 「Download from web or upload from disk」ダイアログの下部にあるPaste/Fetch dataを押す。
- 出現したボックスにデータのURL
https://git.io/JfOagを入力し、“Type (set all):”をzipに変更する。 - Startを押す。
- データの名前を
GSE147507_RawReadCounts_Human.zipに変更する。
- データを解凍する。
- ツールパネルからunzipを選ぶ。
- “input_file”を
GSE147507_RawReadCounts_Human.zipにする。
- 得られたデータはカウントデータである。ファイル名は
Series*_細胞株_感染ウィルス_*となっている。
発現差異検出
- ツールパネルからDESeq2を選び、
- “how”:
Select datasets per level - “Factor” - “1: Factor”中:
- “Specify a factor name”:
virus - “1: Factor level”中:
- “Specify a factor level”:
Mock - “Counts file(s)”:
Series1_NHBE_Mock_*サンプルを選択。ボックス右のBrowse Datasetsボタンを押して、出現したダイアログの“Type to Search”tsvを指定し、目的のファイルを複数選択する。
- “Specify a factor level”:
- “2: Factor level”中:
- “Specify a factor level”:
SARS-CoV-2 - “Counts file(s)”:
Series1_NHBE_SARS-CoV-2_*サンプルを選択
- “Specify a factor level”:
- “Specify a factor name”:
- “Files have header?”:
Yes - “Output normalized counts table”:
Yes - Executeボタンを押して実行する。
- “how”:
- Gene lengthを計算する。
- 上部の Shared Data -> Data Libraries から、共有データにアクセスし、Genomes + annotaions / Annotations / 2017 / hg38_UCSC_07_15_genes.gtf を選んでhistoryにimportする。
- ツールパネルからGene length and GC contentsを選ぶ。
- “Select a built-in GTF file or one from your history”:
Use a GTF from histroy - “Choose history if you don’t see the correct GTF”: 先ほどimportしたGTFファイル
- “Select a built-in FASTA or one from your history”:
Use a built-in FASTA - “Select a FASTA file”:
Human Dec. 2013 (GRCh38/hg38) (hg38)
これ以降の解析はここの「発現差異遺伝子の可視化」以下とほぼ同じである。 ただし、「発現差異遺伝子の機能推定」のgoseqでは、
- “Gene lengths file”: 上で計算した Gene length
- “Select a genome to use”:
Human (hg38) - “Select Gene ID format”:
Gene Symbol
を選ぶ。