次世代シークエンサーを用いた遺伝子発現解析
概要
本課題では、キイロショウジョウバエ Drosophila melanogaster のトランスクリプトーム解析 (Brooks et al. 2011) を題材とし、次世代シークエンサーのデータを用いた遺伝子発現解析を体験する。具体的には以下の実験を行う:
- 二つの条件で得られたRNA-Seqのショートリードを参照ゲノムにマッピングし、各遺伝子の発現量を推定する。
- 二つの条件の間で発現に差異がある遺伝子を同定する。
- これらの解析結果に基づき遺伝子機能解析を行い、二つの条件の生物学的な差異を考察する。
以上の解析は、生命科学のデータを解析するためのウェブ環境Galaxyの上で実行する。これらの問題を解くことにより、次世代シークエンサーが産出するデータを計算機で解析することが生命科学においてどれだけ有効であるかを確認する。
Galaxyの準備
まずアカウントを作成する。
- https://usegalaxy.eu/ を Chrome で開く。
- 右上の Login or Register (あるいは ログイン/登録)から新規アカウントを登録する。
- メールアドレス、パスワード、ニックネームを入力してサブミットする。
- 登録したメールアドレスに確認メールが届くので、それに記載されている確認リンクをクリックすることによってアカウント登録が完了する。
- 登録したメールアドレスとパスワードを使ってログインする。
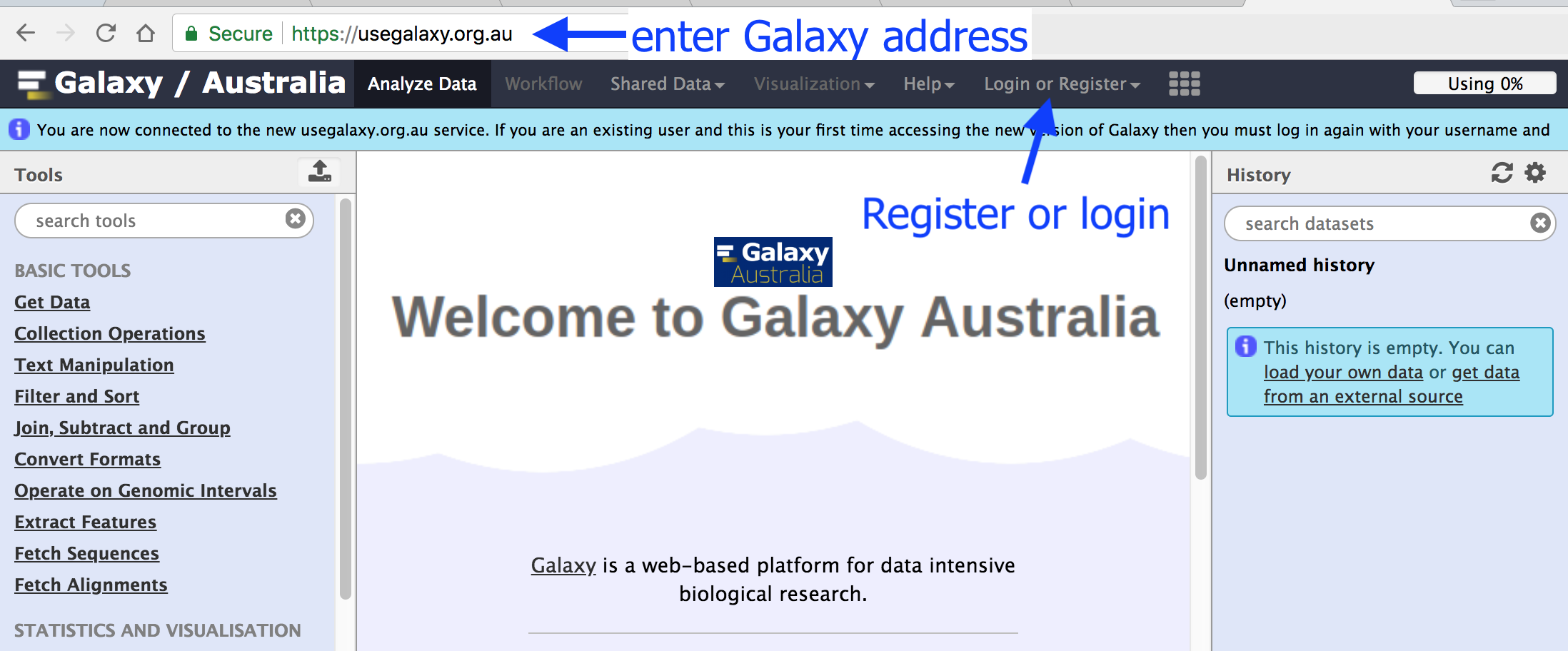
Galaxyの画面インターフェイスは以下のようになっている。
- 左側:ツールパネル
(Galaxyによる解析で使用することができるツールが並んでいる) - 中央:メインパネル
(ツールの実行結果を表示する) - 右側:ヒストリー
(実行した解析の履歴を見ることができる)
前処理
データ取得
-
新しいヒストリーを作成し、「RNA-Seq」という名前を付ける。
- FASTQファイルをインポートする。
- 上部の 共有データ -> データライブラリー から、共有データにアクセスし、 Galaxy courses / RNA-Seq 2018 / raw sequences を選ぶ。
- 以下の4つのファイルを選択し、上部の To History から as Datasetsを選ぶ。
- ヒストリーRNA-Seqを選んでImportする。
サンプル名 ファイル名 GSM461177 (untreated) GSM461177_1, GSM461177_2 GSM461180 (treated) GSM461180_1, GSM461180_2 -
各データセットについて、 Attributesタブ-Nameがファイル名 (e.g. GSM461177_1) 、 Database/Buildが dm6 に、 Datatypesタブ-New Typeが fastqsanger となっていることを確認する。
-
各データセットについて、Tagが#サンプル名 (e.g. #GSM461177) になっていることを確認する。なっていなければ、データセット名をクリックし、Add Tagsをクリックすると変更できる。この際、元のタブは削除する。
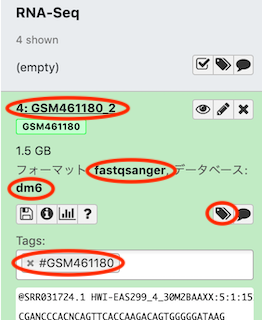
Question
- それぞれのデータセットはどのようなデータ形式か?
品質管理
次世代シークエンサーによるDNA解読では、ある一定の確率(プラットフォームによって異なるが\(10^{-2} \sim 10^{-3}\))で読み取りエラーが発生し、特にシークエンシング後半には高い頻度で読み取りエラーが起こることが知られている。 以降の解析の信頼性を保証するために、最初に品質管理をすることが重要である。 シークエンシングの結果として得られる塩基配列を含むFASTQ形式のファイルには、一塩基ごとのクオリティスコア(推定エラー率)が付与されている。 これをFastQCというツールで可視化することによって、各サンプル対するシークエンシング結果の品質を確認する。
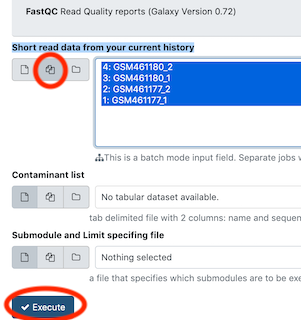
- ツールパネルから FastQC を選ぶ。
- 先頭の“Raw read data from your current history”で”Multiple datasets”ボタンを押して複数のデータセットを選択する。WindowsではCtrlキーを押しながら選択すると複数のデータセットを選択することができる。
- Executeボタンを押して実行する。
- 左側のツールパネルから MultiQC を選ぶ。
- 先頭の“1: results”において
- “Which tool was used generate logs?”:
FastQC - “Insert FastQC Output”
- “Type of FastQC output?”:
Raw data - “FastQC output”: FastQCの出力のうちRaw data (e.g. xx: FastQC on Data xx: RawData) を4つ選ぶ。
- “Which tool was used generate logs?”:
- Run Toolボタンを押して実行する。
- 先頭の“1: results”において
-
出力された結果のうち MultiQC on data xx, data xx, and others: Webpages内の目玉マークをクリックし、4つのデータセットのFastQC結果をまとめた結果を調べる。
Question
- リードの品質は十分と言えるだろうか?
GSM461177_1とGSM461177_2の関係は?
次に、Cutadaptを使って品質が低いリードを除去する。
-
ツールパネルから Cutadapt を選ぶ。
- “Single-end or Paired-end reads?”:
Paired-end- “FASTQ/A file #1”:
_1がついたfastqsangerデータセット2つ - “FASTQ/A file #2”:
_2がついたfastqsangerデータセット2つ
を選ぶ。複数選択する際に、選択する順番に気をつけること。
- “FASTQ/A file #1”:
-
“Filter Options”の中の“Minimum length”:
20 -
“Read Modification Options”の中の“Quality cutoff”:
20 -
“Outputs selector”の中の“Report” にチェック
- Run Toolボタンを押して実行する。
- “Single-end or Paired-end reads?”:
-
Cutadaptの結果 (xx: Cutadapt on Data xx and Data xx: Report) を調べる。
Question
- それぞれのデータセットで低品質のための削除された塩基の数は?
- それぞれのデータセットで短くなりすぎたために削除されたリード数は?
マッピング
ショートリードをD. melanogasterの参照ゲノムdm6にマッピングする。 mRNAのシークエンシングでは成熟mRNAを逆転写したcDNAを読み取る。 そのため、参照ゲノムにリードをマッピングするとイントロンに相当する部位がスキップされ、下図のようにスプライス部位で分割されるリードが観測される。

本課題では、スプライシングに対応したアラインメントツールであるSTAR Alignerを用いてRNA-Seqデータを参照ゲノムにマッピングする。
遺伝子アノテーションファイルの取得
- 上部の 共有データ -> データライブラリ から、共有データにアクセスし、 Galaxy courses / RNA-Seq 2018 / annotation を選ぶ。
- Drosophila_melanogaster.BDGP6.87.gtfを選ぶ。
- ヒストリーRNA-Seqを選んでImportする。
- 取得したデータのFormatが
gtf, Databaseがdm6であることを確認する。
マッピングを実行
-
ツールパネルからRNA STARを選ぶ。
- “Single-end or paired-end reads”:
Paired-end (as individual datasets)- “RNA-Seq FASTQ/FASTA file, forward reads”: Cutadaptの出力のうちRead 1 (e.g. xx: Cutadapt on Data xx and Data xx: Read 1 Output)
- “RNA-Seq FASTQ/FASTA file, reverse reads”: Cutadaptの出力のうちRead 2 (e.g. xx: Cutadapt on Data xx and Data xx: Read 2 Output)
を選ぶ。複数選択する際に、選択する順番に気をつけること。
- “Custom or built-in reference genome”:
Use a built-in index- “Reference genome with or without an annotation”:
use genome reference without builtin gene-model but provide a gtf - “Select reference genome”:
Fly (Drosophila Melanogaster): dm6 Full - “Gene model (gff3,gtf) file for splice junctions”:
Drosophila_melanogaster.BDGP6.87.gtf - “Length of the genomic sequence around annotated junctions”:
36
(この値はリード長-1にする必要がある)
- “Reference genome with or without an annotation”:
- Run Toolボタンを押して実行する。
マッピング結果をまとめる
- ツールパネルからMultiQCを選ぶ。
- “Which tool was used generate logs?”:
STAR - “Insert STAR output“をクリック
- “Type of STAR output?”:
Log - “STAR Log output”: STARのLog (e.g. xx: RNA STAR on data xx, data xx, and data xx: log)を2つ選ぶ。
- Run Toolボタンを押して実行する。
Question
- マッピング率(マップされたリードの割合)はそれぞれのサンプルで何%か?
- マッピングに失敗したリードはどのような理由でマッピングに失敗したのだろうか?
- STARの出力はどのような形式のファイルだろうか?どのような情報が含まれているか?
マッピング結果の可視化
マッピングの結果はゲノムブラウザIGVを用いて可視化する。
- IGVをここからダウンロードする。使用しているコンピュータにJavaがインストールされていない場合には “Java included” と書いてあるものを選ぶ。
- ダウンロードしたZIPファイルを解凍して、中にある
igv.bat(Windows) かigv.command(Mac) を実行する。 - IGVのメニュー Genomes->Load Genomes from Serverから
D. melanogaster (dm6)を選択し、ゲノム・遺伝子アノテーションをダウンロードする。 - Galaxyに戻り、STARの実行結果として出力される
bamファイル (e.g. xx: RNA STAR on data xx, data xx, and data xx: mapped.bam) を開き、display with IGVのlocalをクリックする。
- IGVのアドレス入力欄に
chr4:540,000-560,000を入力し、この領域を拡大表示する。 - IGVのBAM fileで右クリックし、Sashimi plotを選択する。
Question
- グレーのピークは何を意味しているか?
- マッピングされたリードをつなぐ線は何を意味しているか?
- Sashimi plotで円弧につけられた数字は何を意味しているか?
- Sashimi plotの下部に見える青いボックス群は何を意味しているか?
遺伝子発現差異解析
RNA-Seqリードを参照ゲノムにマッピングした結果から、遺伝子領域にマッピングされたリードの本数を計算し、各遺伝子の発現量を推定する。 複数の条件(treated条件3サンプルとuntreated条件4サンプル)における遺伝子発現量を比較することによって、発現に差異がある遺伝子を同定する。 本課題では、遺伝子ごとにマッピングされたリードの本数を計量するためにfeatureCountsを使用し、 発現差異をDESeq2を用いて同定する。 発現差異解析の結果は、同定された発現差異遺伝子のリストと概要を図示したもの(主成分分析やクラスタリング)として得られる。
ストランドネスを判定
IlluminaのRNA-SeqではRNAをcDNAに逆転写してからPCR増幅を行うため、遺伝子がゲノムの正鎖、逆鎖のどちらにあるかに関わらず、リードはどちらにもマッピングされる (unstranded RNA-Seq)。 そのため、RNAがどちらの鎖から発現しているかを区別することができない。 一方、特別な処理 (e.g. dUTP tagging) を施すことによってどちらの鎖から発現しているかを区別することが可能となる (stranded RNA-Seq)。 どちらの手法を使ってRNA-Seqを行なっているかによって遺伝子発現量に差が現れるため、この実験がunstranded RNA-Seqかstranded RNA-Seqかを知る必要がある。
- ツールパネルからConvert GTF to BED12を選び、
- GTF File to convertをDrosophila_melanogaster.BDGP6.87.gtfとして
- Run Toolボタンを押す。
- ツールパネルからInfer Experimentを選び、
- “Input .bam file”: RNA STARの出力mapped.bam (e.g. xx: RNA STAR on data xx, data xx, and data xx: mapped.bam)
- “Reference gene model”: Convert GTF to BED12の出力 (e.g. xx: GTF to BED12 on data xx)
- “Number of reads sampled from SAM/BAM file (default = 200000)”:
200000 - Run Toolボタンを押して実行する。
Question
- このデータは unstranded か stranded か?
リードカウント
- ツールパネルからfeatureCountsを選び、
- “Alignment file”: RNA STARの出力mapped.bam (e.g. xx: RNA STAR on data xx, data xx, and data xx: mapped.bam)
- “Specify strand information”:
Unstranded - “Gene annotation file”:
A GFF/GTF file in your history- “Gene annotation file”:
Drosophila_melanogaster.BDGP6.87.gtf - “GFF feature type filter”:
exon - “GFF gene identifier”:
gene_id
- “Gene annotation file”:
- “Output format”:
Gene-ID "\t" read-count (MultiQC/DESeq2/edgeR/limma-voom compatible) - “Create gene-length file”:
Yes - “Does the input have read pairs?”:
Yes, paired-end but still count them as if individual reads - “Read filtering options”中の
- “Minimum mapping quality per read”:
10
- “Minimum mapping quality per read”:
- “Advanced options”中の
- “Allow read to contribute to multiple features”:
Disabled
- “Allow read to contribute to multiple features”:
- Run Toolボタンを押して実行する。
- ツールパネルからMultiQCを選び、
- “Results”中の
- “Which tool was used generate logs?”:
featureCounts - “Output of FeatureCounts”: featureCounts toolの出力 (e.g. xx: featureCounts on data xx and data xx: Summary) の二つを選択
- “Which tool was used generate logs?”:
- Run Toolボタンを押して実行する。
- “Results”中の
Question
- どのぐらいのリードが遺伝子に割り当てられたか?
発現差異検出
- 7サンプルに対して事前に計算されたfeatureCountsの結果をインポートする。
- 上部の Shared Data -> Data Libraries から、共有データにアクセスし、 Galaxy courses / RNA-Seq 2018 / gene_counts を選ぶ。
- すべてのファイルを選択し、上部の To History から as Datasetsを選ぶ。
GSM461176_untreat_single.countsGSM461177_untreat_paired.countsGSM461178_untreat_paired.countsGSM461179_treat_single.countsGSM461180_treat_paired.countsGSM461181_treat_paired.countsGSM461182_untreat_single.counts
- ヒストリーRNA-Seqを選んでImportする。
- ツールパネルからDESeq2を選び、
Select datasets per level- “Factor” - “1: Factor”中:
- “Specify a factor name”:
Treatment - “Factor level” - “1: Factor level”中:
- “Specify a factor level”:
treated - “Counts file(s)”: “treat”サンプルの3つを選択
- “Specify a factor level”:
- “Factor” - “2: Factor level”:
- “Specify a factor level”:
untreated - “Counts file(s)”: “untreat”サンプルの4つを選択
- “Specify a factor level”:
- “Specify a factor name”:
- “Insert Factor” (“Insert Factor level”ではなく) をクリック
- “2: Factor”中:
- “Specify a factor name”:
Sequencing - “1: Factor level”中:
- “Specify a factor level”:
PE - “Counts file(s)”: paired endサンプルの4つを選択
- “Specify a factor level”:
- “Insert Factor level”をクリック
- “2: Factor level”中:
- “Specify a factor level”:
SE - “Counts file(s)”: single endサンプルの3つを選択
- “Specify a factor level”:
- “Specify a factor name”:
- “Files have header?”:
No - “Output options”:
Generate plots for visualizing the analysis resultsとOutput normalised countsをチェック - Run Toolボタンを押して実行する。
Question
- PCA(DESeq2 plotsの一枚目)において
- PC1を特徴づけているものは何か?
- PC2を特徴づけているものは何か?
- ヒートマップ(DESeq2 plotsの二枚目)におけるクラスタリングでは、どのような基準でグルーピングされているか?
発現差異遺伝子の可視化
- ツールパネルからFilter data on any column using simple expressions を選び、
- “Filter”: DESeq2のresult file (e.g. xx: DESeq2 result file on data 7, data 4, and others)
- “With following condition”:
c7<0.05 - Run Toolボタンを押して実行する。
- ツールパネルからFilter data on any column using simple expressionsを選び、
- “Filter”: 1.の結果 (e.g. xx: Filter on data xx)
- “With following condition”:
abs(c3)>1 - Run Toolボタンを押して実行する。
Question
- 統計的に有意(p値<0.05)な発現変動を示している遺伝子はいくつか?
- その中でも変動が特に大きい(log2(Fold Change)>1)遺伝子はいくつか?
- コピー&ペーストでヘッダーを作成
- 最後のFilterの結果を表示し、1行目のヘッダーをコピーする。
- ツールパネル上部にあるUpload dataから
- Paste/Fetch dataを選択、
- 1.でコピーした内容を中央にペースト
- Typeをtabularに変更し、Start
- ツールパネルからConcatenate datasetsを選び、
Pasted entryを選択- Insert Datasetをクリック
- 最後のFilter出力を選択
- Run Toolボタンをクリック
- ツールパネルからJoin two Datasetsを選び、
- “Join”: DESeq2の出力normalized counts file (e.g. xx: Normalized counts file on data xx, data xx, others)
- “using column”:
Column: 1 - “with”: Concatenate datasetsの出力 (e.g. xx: Concatenate datasets on data xx and data xx)
- “and column”:
Column: 1 - “Keep lines of first input that do not join with second input”:
No - “Keep the header lines”:
Yes - Run Toolボタンをクリック
- ツールパネルからCutを選び、
- “Cut columns”:
c1-c8 - “Delimited by”:
Tab - “From”: Joinの出力 (e.g. xx: Join two Datasets on data xx and data xx)
- Run Toolボタンをクリック
- “Cut columns”:
- ツールパネルからheatmap2を選び、
- “Input should have column headers”: Cutの結果 (e.g. xx: Cut on data xx)
- “Data transformation”:
"Log2(value+1) transform my data - “Enable data clustering”:
Yes - “Labeling columns and rows”:
Label columns and not rows - “Type of colormap to use”:
Gradient with 3 colors - Run Toolボタンをクリック
Question
- 縦軸と横軸においてどのような基準でクラスタリングされているか?
発現差異遺伝子の機能推定
発現差異遺伝子は、遺伝子オントロジー (Gene Ontology; GO) 解析によって解釈を与えることができる。 GOとは、遺伝子の生物的プロセス、細胞の構成要素および分子機能に着目し、遺伝子ごとに階層的に付けられたアノテーションのことである。 発現差異遺伝子群において有意に多く現れているGOを検出することができれば、それが条件間の生物学的な差異と関連があると考えることができる。 本課題では、goseqを用いてGO解析を行う。
- ツールパネルからCompute an expression on every rowを選び、上部”versions”(Run toolボタンの左側)よりversion 1.6を選択、
- “Add expression”:
bool(c7<0.05) - “as a new column to”: DESeq2のresult file (e.g. xx: DESeq2 result file on data xx, data xx, and others)
- Run Toolボタンをクリック
- “Add expression”:
- ツールパネルからCutを選び、
- “Cut columns”:
c1,c8 - “Delimited by”:
Tab - “From”: Computeの出力 (e.g. xx: Compute on data 8)
- Run Toolボタンをクリック
- “Cut columns”:
- ツールパネルからChange Caseを選び、
- “From”: Cutの出力 (xx: Cut on data xx)
- “Change case of columns”:
c1 - “Delimited by”:
Tab - “To”:
Upper case - Run Toolボタンをクリック
- ツールパネルからChange Caseを選び、
- “From”: feature lengthの出力 (e.g. xx: featureCounts on data xx, data xx: Feature lengths)
- “Change case of columns”:
c1 - “Delimited by”:
Tab - “To”:
Upper case - Run Toolボタンをクリック
- ツールパネルからgoseqを選び、
- “Differentially expressed genes file”: Change Caseで作成したファイルの一つ目
- “Gene lengths file”: Change Caseで作成したファイルの二つ目
- “Gene categories”:
Get categories - “Select a genome to use”:
Fruit fly (dm6) - “Select Gene ID format”:
Ensembl Gene ID - “Select one or more categories”:
GO: Cellular Component,GO: Biological Process,GO: Molecular Function - Run Toolボタンをクリック
Question
- over-representedあるいはunder-represented (補正P値 < 0.05) なGO termはそれぞれいくつずつか?
- GO termのカテゴリ (MF: Molecular Function, CC: Cellular Component, BP: Biological Process) について over-represented, under-representedなGO termはいくつずつか?
レポートの書き方
「1. 次世代シークエンサーを用いた遺伝子発現解析」については、以下の項目についてレポートをまとめてください。
-
目的
本実験の目的を簡潔に書く。 -
手法
各ステップでの内容や目的を含めて時系列で書く。特に以下に挙げたツールは用途や特徴を簡潔に書く。- FastQC
- Cutadapt
- RNA STAR
- featureCounts
- DESeq2
- heatmap2
- goseq
-
結果
各ステップについて、適宜結果の図を貼り付けつつ実行結果を記述する。「Question」の解答もここに記載する。 - 考察
各自、考察を行う。以下は考察の例である。- 各条件で異なるスプライシングが観測される遺伝子を見つけ、その効果について考察する。
- 発現差異が観測された遺伝子にはどのような共通点があるかを考察する。
- 元の論文を参考に、Pasilla遺伝子とその哺乳類におけるオルソログ遺伝子NOVA1およびNOVA2について、その制御対象となる遺伝子が進化的に保存されているかどうかを解析する。
-
結論
本課題の結論を簡潔に書く。 - 参考資料
引用・利用したものがあればここに記載する。
発展課題
現在、COVID-19関連のバイオインフォマティクス解析が盛んに行われている。 こちらのサイトでは、Galaxyを用いてCOVID-19のゲノム解析、進化解析、ドッキング解析を行う例が示されている。 また、ここにCOVID-19感染細胞のトランスクリプトーム解析の例を示した。 これらを参考に、各自オリジナルの解析を行ってください。
発展課題をやった場合には、レポートの枚数は +4 枚まで認めます。
参考文献
- Galaxy Training - Transcriptomics - Reference-based RNA-Seq data analysis (この演習は基本的にここの内容に沿ったものとなっています。)